Data Preparation¶
Displaying large amounts of data often requires first turning it into not-so-large amounts of data. Clodius is a program and library designed to aggregate large datasets to make them easy to display at different resolutions.
Bed Files¶
BED files specify genomic intervals. They are aggregated according to an importance function that determines which values should be visible at lower zoom levels. This importance function is user specified. In the absence of any clear ranking of the different lines in the BED file, a random value can be used in lieu of the importance function.
Example BED file:
chr9 135766734 135820020 TSC1 Biallelic inactivation may predict sensitivity to MTOR inhibitors
chr16 2097895 2138721 TSC2 Biallelic inactivation may predict sensitivity to MTOR inhibitors
chr3 10183318 10195354 VHL May signal the presence of a germline mutation.
chr11 32409321 32457081 WT1 May signal the presence of a germline mutation.
This file can be aggregated like so:
clodius aggregate bedfile \
--assembly hg19 \
short.bed
And then imported into higlass after copying to the docker temp directory (cp short.bed.multires ~/hg-tmp/):
docker exec higlass-container python \
higlass-server/manage.py ingest_tileset \
--filename /tmp/short.bed.multires \
--filetype beddb \
--datatype bedlike \
--coordSystem b37
Bedpe-like Files¶
BEDPE-like files contain two sets of chromosomal coordinates:
chr10 74160000 74720000 chr10 74165000 74725000
chr12 120920000 121640000 chr12 120925000 121645000
chr15 86360000 88840000 chr15 86365000 88845000
To view such files in HiGlass, they have to be aggregated so that tiles don’t contain too many values and slow down the renderer:
clodius aggregate bedpe \
--assembly hg19 \
--chr1-col 1 --from1-col 2 --to1-col 3 \
--chr2-col 4 --from2-col 5 --to2-col 6 \
--output-file domains.txt.multires \
domains.txt
This requires the –chr1-col, –from1-col, –to1-col, –chr2-col, –from2-col, –to2-col parameters to specify which columns in the datafile describe the x-extent and y-extent of the region.
The priority with which regions are included in lower resolution tiles is specified by the –impotance-column parameter. This can either provide a value, contain random, or if it’s not specified, default to the size of the region.
BedGraph files¶
Warning
The order of the chromosomes in the bedgraph file have to be consistent with the order specified for the assembly in the negspy repository.
Ordering the chromosomes in the input file¶
input_file=~/Downloads/phastCons100way.txt.gz;
output_file=~/Downloads/phastConst100way_ordered.txt;
chromnames=$(awk '{print $1}' ~/projects/negspy/negspy/data/hg19/chromInfo.txt);
for chr in $chromnames;
do echo ${chr};
zcat $input_file | grep "\t${chr}\t" >> $output_file;
done;
Aggregation by addition¶
Assume we have an input file that has id chr start end value1 value2 pairs:
location chrom start end copynumber segmented
1:2900001-3000000 1 2900001 3000000 -0.614 -0.495
1:3000001-3100000 1 3000001 3100000 -0.407 -0.495
1:3100001-3200000 1 3100001 3200000 -0.428 -0.495
1:3200001-3300000 1 3200001 3300000 -0.437 -0.495
We can aggregate this file by recursively summing adjacent values. We have to
indicate which column corresponds to the chromosome (--chromosome-col 2),
the start position (--from-pos-col 3), the end position (--to-pos-col 4)
and the value column (--value-col 5). We specify that the first line
of the data file contains a header using the (--has-header) option.
clodius aggregate bedgraph \
test/sample_data/cnvs_hw.tsv \
--output-file ~/tmp/cnvs_hw.hitile \
--chromosome-col 2 \
--from-pos-col 3 \
--to-pos-col 4 \
--value-col 5 \
--assembly grch37 \
--nan-value NA \
--transform exp2 \
--has-header
Data Transform¶
The dataset used in this example contains copy number data that has been log2
transformed. That is, the copy number given for each bin is the log2 of the
computed value. This is a problem for HiGlass’s default aggregation method of
summing adjacent values since 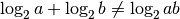 .
.
Using the --transform exp2 option tells clodius to raise two to the
power of the provided value before doing the transformation and storing. As
an added benefit, NaN values become apparent in the resulting because they
have values of 0.
NaN Value Identification¶
NaN (not a number) values in the input file can be specified using the
--nan-value option. For example, --nan-value NA indicates that
whenever NA is encountered as a value it should be treated as NaN. In the
current implementation, NaN values are simply treated as 0. In the future, they
should be assigned a special value so that they are ignored by HiGlass.
When NaN values are aggregated by summing, they are treated as 0 when added to another number. When two NaN values are added to each other, however, the result is Nan.
NaN Value Counting¶
Sometimes, we just want to count the number of NaN values in the file. The
--count-nan option effectively treats NaN values as 1 and all other values
as 0. This makes it possible to display a track showing how many NaN values are
present in each interval. It also makes it possible to create compound tracks
which use that information to normalize track values.
bigWig files¶
bigWig files store genomic data in a compressed, indexed form that allows rapid retrieval and visualization. bigWig files can be loaded directly into HiGlass using the vector datatype and bigwig filetype:
docker exec higlass-container python \
higlass-server/manage.py ingest_tileset \
--filename /tmp/cnvs_hw.bigWig \
--filetype bigwig \
--datatype vector \
--coordSystem hg19
Important: BigWig files have to be associated with a chromosome order!!
This means that there needs to be a chromsizes file for the
specified assembly (coordSystem) in the higlass database. If no coordSystem
is specified for the bigWig file in ingest_tileset, HiGlass will try to
find one in the database that matches the chromosomes present in the bigWig file.
If a chromsizes tileset is found, it’s coordSystem will also be used for
the bigWig file. If none are found, the import will fail. If more than one is found,
the import will also fail. If a coordSystem is specified for the bigWig, but no
chromsizes are found on the server, the import will fail.
TLDR: The simplest way to import a bigWig is to have a chromsizes present e.g.
ingest_tileset --filetype chromsizes-tsv --datatype chromsizes --coordSystem hg19 chromSizes.tsvand then to add the bigWig with the same coordSystem:
ingest_tileset --filetype bigwig --datatype vector --coordSystem hg19 chromSizes.tsvGene Annotation Tracks¶
HiGlass uses a specialized track for displaying gene annotations. It is rougly based on UCSC’s refGene files (e.g. http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/). For any identifiable genome assembly the following commands can be run to generate a list of gene annotation that can be loaded as a zoomable track in HiGlass.
Prerequisites¶
For any assembly, there needs to a refGene file:
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/refGene.txt.gz
And a list of chromosome sizes in the negspy python package.
If there are no available chromosome sizes for this assembly in negspy, adding them is simply a matter of downloading the list from UCSC (e.g. http://hgdownload.cse.ucsc.edu/goldenpath/hg19/bigZips/hg19.chrom.sizes)
Set the assembly name and species ID¶
ASSEMBLY=mm9
TAXID=10090
#ASSEMBLY=hg19
#TAXID=9606
#ASSEMBLY=sacCer3
#TAXID=559292
#ASSEMBLY=dm6
#TAXID=7227
Download data from UCSC and NCBI¶
mkdir -p ~/data/genbank-data/${ASSEMBLY}
wget -N -P ~/data/genbank-data/${ASSEMBLY}/ \
http://hgdownload.cse.ucsc.edu/goldenPath/${ASSEMBLY}/database/refGene.txt.gz
wget -N -P ~/data/genbank-data/ \
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2refseq.gz
wget -N -P ~/data/genbank-data/ \
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene_info.gz
wget -N -P ~/data/genbank-data/ \
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2pubmed.gz
Preprocess data¶
# remove entries to chr6_...
gzcat ~/data/genbank-data/${ASSEMBLY}/refGene.txt.gz \
| awk -F $'\t' '{if (!($3 ~ /_/)) print;}' \
| sort -k2 > ~/data/genbank-data/${ASSEMBLY}/sorted_refGene
wc -l ~/data/genbank-data/${ASSEMBLY}/sorted_refGene
zgrep ^${TAXID} ~/data/genbank-data/gene2refseq.gz \
> ~/data/genbank-data/${ASSEMBLY}/gene2refseq
head ~/data/genbank-data/${ASSEMBLY}/gene2refseq
zgrep ^${TAXID} ~/data/genbank-data/gene_info.gz \
| sort -k 2 \
> ~/data/genbank-data/${ASSEMBLY}/gene_info
head ~/data/genbank-data/${ASSEMBLY}/gene_info
zgrep ^${TAXID} ~/data/genbank-data/gene2pubmed.gz \
> ~/data/genbank-data/${ASSEMBLY}/gene2pubmed
head ~/data/genbank-data/${ASSEMBLY}/gene2pubmed
# awk '{print $2}' ~/data/genbank-data/hg19/gene_info \
# | xargs python scripts/gene_info_by_id.py \
# | tee ~/data/genbank-data/hg19/gene_summaries.tsv
# output -> geneid \t citation_count
Processing¶
cat ~/data/genbank-data/${ASSEMBLY}/gene2pubmed \
| awk '{print $2}' \
| sort \
| uniq -c \
| awk '{print $2 "\t" $1}' \
| sort \
> ~/data/genbank-data/${ASSEMBLY}/gene2pubmed-count
head ~/data/genbank-data/${ASSEMBLY}/gene2pubmed-count
# output -> geneid \t refseq_id
cat ~/data/genbank-data/${ASSEMBLY}/gene2refseq \
| awk -F $'\t' '{ split($4,a,"."); if (a[1] != "-") print $2 "\t" a[1];}' \
| sort \
| uniq \
> ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid
head ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid
wc -l ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid
#output -> geneid \t refseq_id \t citation_count
join ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid \
~/data/genbank-data/${ASSEMBLY}/gene2pubmed-count \
| sort -k2 \
> ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid_count
head ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid_count
wc -l ~/data/genbank-data/${ASSEMBLY}/geneid_refseqid_count
# output -> geneid \t refseq_id \t chr (5) \t strand(6) \t txStart(7) \t txEnd(8) \t cdsStart(9) \t cdsEnd (10) \t exonCount(11) \t exonStarts(12) \t exonEnds(13)
join -1 2 -2 2 \
~/data/genbank-data/${ASSEMBLY}/geneid_refseqid_count \
~/data/genbank-data/${ASSEMBLY}/sorted_refGene \
| awk '{ print $2 "\t" $1 "\t" $5 "\t" $6 "\t" $7 "\t" $8 "\t" $9 "\t" $10 "\t" $11 "\t" $12 "\t" $13 "\t" $3; }' \
| sort -k1 \
> ~/data/genbank-data/${ASSEMBLY}/geneid_refGene_count
head ~/data/genbank-data/${ASSEMBLY}/geneid_refGene_count
wc -l ~/data/genbank-data/${ASSEMBLY}/geneid_refGene_count
# output -> geneid \t symbol \t gene_type \t name \t citation_count
join -1 2 -2 1 -t $'\t' \
~/data/genbank-data/${ASSEMBLY}/gene_info \
~/data/genbank-data/${ASSEMBLY}/gene2pubmed-count \
| awk -F $'\t' '{print $1 "\t" $3 "\t" $10 "\t" $12 "\t" $16}' \
| sort -k1 \
> ~/data/genbank-data/${ASSEMBLY}/gene_subinfo_citation_count
head ~/data/genbank-data/${ASSEMBLY}/gene_subinfo_citation_count
wc -l ~/data/genbank-data/${ASSEMBLY}/gene_subinfo_citation_count
# 1: chr (chr1)
# 2: txStart (52301201) [9]
# 3: txEnd (52317145) [10]
# 4: geneName (ACVRL1) [2]
# 5: citationCount (123) [16]
# 6: strand (+) [8]
# 7: refseqId (NM_000020)
# 8: geneId (94) [1]
# 9: geneType (protein-coding)
# 10: geneDesc (activin A receptor type II-like 1)
# 11: cdsStart (52306258)
# 12: cdsEnd (52314677)
# 14: exonStarts (52301201,52306253,52306882,52307342,52307757,52308222,52309008,52309819,52312768,52314542,)
# 15: exonEnds (52301479,52306319,52307134,52307554,52307857,52308369,52309284,52310017,52312899,52317145,)
join -t $'\t' \
~/data/genbank-data/${ASSEMBLY}/gene_subinfo_citation_count \
~/data/genbank-data/${ASSEMBLY}/geneid_refGene_count \
| awk -F $'\t' '{print $7 "\t" $9 "\t" $10 "\t" $2 "\t" $16 "\t" $8 "\t" $6 "\t" $1 "\t" $3 "\t" $4 "\t" $11 "\t" $12 "\t" $14 "\t" $15}' \
> ~/data/genbank-data/${ASSEMBLY}/geneAnnotations.bed
head ~/data/genbank-data/${ASSEMBLY}/geneAnnotations.bed
wc -l ~/data/genbank-data/${ASSEMBLY}/geneAnnotations.bed
python scripts/exonU.py \
~/data/genbank-data/${ASSEMBLY}/geneAnnotations.bed \
> ~/data/genbank-data/${ASSEMBLY}/geneAnnotationsExonUnions.bed
wc -l ~/data/genbank-data/${ASSEMBLY}/geneAnnotationsExonUnions.bed
Creating a HiGlass Track¶
workon hg-server
ASSEMBLY=mm9
clodius aggregate bedfile \
--max-per-tile 20 --importance-column 5 \
--assembly ${ASSEMBLY} \
--output-file ~/data/tiled-data/gene-annotations-${ASSEMBLY}.db \
--delimiter $'\t' \
~/data/genbank-data/${ASSEMBLY}/geneAnnotationsExonUnions.bed
aws s3 cp ~/data/tiled-data/gene-annotations-${ASSEMBLY}.db \
s3://pkerp/public/hg-server/data/${ASSEMBLY}/
Importing into HiGlass¶
curl -u `cat ~/.higlass-server-login` \
-F "datafile=@/Users/peter/data/tiled-data/gene-annotations-${ASSEMBLY}.db" \
-F "name=Gene Annotations (${ASSEMBLY})" \
-F 'filetype=beddb' \
-F 'datatype=gene-annotation' \
-F 'coordSystem=${ASSEMBLY}' \
-F 'coordSystem2=${ASSEMBLY}' \
http://higlass.io:80/api/v1/tilesets/
Chromosomes¶
curl -u `cat ~/.higlass-server-login` \
-F "datafile=@/Users/peter/tmp/chromSizes_hg38.tsv" \
-F "name=Chromosomes (hg38)" \
-F 'filetype=chromsizes-tsv' \
-F 'datatype=chromsizes' \
-F "coordSystem=${ASSEMBLY}" \
-F "coordSystem2=${ASSEMBLY}" \
http://higlass.io:80/api/v1/tilesets/
Hitile files¶
Hitile files are HDF5-based 1D vector files containing data at multiple resolutions.
To see hitile datasets in higlass, use the docker container to load them:
docker exec higlass-container python \
higlass-server/manage.py ingest_tileset \
--filename /tmp/cnvs_hw.hitile \
--filetype hitile \
--datatype vector
Point your browser at 127.0.0.1:8989 (or wherever it is hosted), click on the little ‘plus’ icon in the view and select the top position. You will see a listing of available tracks that can be loaded. Select the dataset and then choose the plot type to display it as.
Cooler files¶
Cooler files (extension .cool) store arbitrarily large 2D genomic matrices, such as those produced via Hi-C and other high throughput proximity ligation experiments. HiGlass can render cooler files containing matrices of the same dataset at a range of bin resolutions or zoom levels, so called multiresolution cool files (typically denoted .mcool).
From pairs¶
Note
Starting with cooler 0.7.9, input pairs data no longer needs to be sorted and indexed.
Often you will start with a list of pairs (e.g. contacts, interactions) that need to be aggregated.
For example, the 4DN-DCIC developed a standard pairs format for HiC-like data. In general, you
only need a tab-delimited file with columns representing chrom1, pos1, chrom2, pos2, optionally gzipped. In the case of Hi-C, these would correspond to the mapped locations of the two ends of a Hi-C ligation product.
You also need to provide a list of chromosomes in semantic order (chr1, chr2, …, chrX, chrY, …) in a two-column chromsizes file.
Ingesting pairs is done using the cooler cload command. Choose the appropriate loading subcommand. If you pairs file is sorted and indexed with pairix or with tabix, use cooler cload pairix or cooler cload tabix, respectively. Otherwise, you can use the new cooler cload pairs command.
Raw pairs example
If you have a raw pairs file or you can stream your data in such a way, you only need to specify the columns that correspond to chrom1, chrom2, pos1 and pos2. For example, if chrom1 and pos1 are the first two columns, and chrom2 and pos2 are in columns 4 and 5, the following command will aggregate the input pairs at 1kb:
cooler cload pairs -c1 1 -p1 2 -c2 4 -p2 5 \
hg19.chrom.sizes:1000 \
mypairs.txt \
mycooler.1000.cool
To pipe in a stream, replace the pairs path above with a dash -.
Note
The syntax <chromsizes_path>:<binsize_in_bp> is a shortcut to specify the genomic bin segmentation used to aggregate the pairs. Alternatively, you can pass in the path to a 3-column BED file of bins.
Indexed pairs example
If you want to create a sorted and indexed pairs file, follow this example. Because an index provides random access to the pairs, this method can be more efficient and parallelized.
cooler csort -c1 1 -p1 2 -c2 4 -p2 5 mypairs.txt hg19.chrom.sizes
will generate a sorted and compressed pairs file mypairs.blksrt.txt.gz along with a companion pairix .px2 index file. To aggregate, use the cload pairix command.
cooler cload pairix hg19.chrom.sizes:1000 mypairs.blksrt.txt.gz mycooler.1000.cool
The output mycooler.1000.cool will serve as the base resolution for the multires cooler you will generate.
From a matrix¶
If your base resolution data is already aggregated, you can ingest data in one of two formats. Use cooler load to ingest.
Note
Prior to cooler 0.7.9, input BG2 files needed to be sorted and indexed. This is no longer the case.
- COO: Sparse matrix upper triangle coordinate list , i.e. tab-delimited sparse matrix triples (
row_id,col_id,count). This is an output of pipelines like HiCPro.
cooler load -f coo hg19.chrom.sizes:1000 mymatrix.1kb.coo.txt mycooler.1000.cool
- BG2: A 2D “extension” of the bedGraph format. Tab delimited with columns representing
chrom1,start1,end1,chrom2,start2,end2, andcount.
cooler load -f bg2 hg19.chrom.sizes:1000 mymatrix.1kb.bg2.gz mycooler.1000.cool
Zoomify¶
To recursively aggregate your matrix into a multires file, use the zoomify command.
cooler zoomify mycooler.1000.cool
The output will be a file called mycooler.1000.mcool with zoom levels increasing by factors of 2. You can also
request an explicit list of resolutions, as long as they can be obtained via integer multiples starting from the base resolution. HiGlass performs well as long as zoom levels don’t differ in resolution by greater than a factor of ~5.
cooler zoomify -r 5000,10000,25000,50000,100000,500000,1000000 mycooler.1000.cool
If this is Hi-C data or similar, you probably want to apply iterative correction (i.e. matrix balancing normalization) by including the --balance option.
Loading pre-zoomed data¶
If the matrices for the resolutions you wish to visualize are already available, you can ingest each one independently into the right location inside the file using the Cooler URI :: syntax.
HiGlass expects each zoom level to be stored at a location named resolutions/{binsize}.
cooler load -f bg2 hg19.chrom.sizes:1000 mymatrix.1kb.bg2 mycooler.mcool::resolutions/1000
cooler load -f bg2 hg19.chrom.sizes:5000 mymatrix.5kb.bg2 mycooler.mcool::resolutions/5000
cooler load -f bg2 hg19.chrom.sizes:10000 mymatrix.10kb.bg2 mycooler.mcool::resolutions/10000
...
See also
See the cooler docs for more information.
You can also type -h or --help after any cooler command for a detailed description.
Multivec Files¶
Multivec files store arrays of arrays organized by chromosome. To aggregate this data, we need an input file where chromsome is a separate dataset. Example:
f = h5py.File('/tmp/blah.h5', 'w')
d = f.create_dataset('chr1', (10000,5), compression='gzip')
d[:] = np.random.random((10000,5))
f.close()
This can be aggregated to multiple resolutions using clodius aggregate multivec:
clodius aggregate multivec \
--chromsizes-filename ~/projects/negspy/negspy/data/hg38/chromInfo.txt \
--starting-resolution 1000 \
--row-infos-filename ~/Downloads/sampled_info.txt \
my_file_genome_wide_hg38_v2.multivec
The –chromsizes-filename option lists the chromosomes that are in the input file and their sizes. The –starting-resolution option indicates that the base resolution for the input data is 1000 base pairs.
Epilogos Data (multivec)¶
Epilogos (https://epilogos.altiusinstitute.org/) show the distribution of chromatin states over a set of experimental conditions (e.g. cell lines). The data consist of positions and states:
chr1 10000 10200 id:1,qcat:[ [-0.2833,15], [-0.04748,5], [-0.008465,7], [0,2], [0,3], [0,4], [0,6], [0,10], [0,11], [0,12], [0,13], [0,14], [0.0006647,1], [0.436,8], [1.921,9] ]
chr1 10200 10400 id:2,qcat:[ [-0.2833,15], [-0.04748,5], [0,3], [0,4], [0,6], [0,7], [0,10], [0,11], [0,12], [0,13], [0,14], [0.0006647,1], [0.004089,2], [0.8141,8], [1.706,9] ]
chr1 10400 10600 id:3,qcat:[ [-0.2588,15], [-0.04063,5], [0,2], [0,3], [0,4], [0,6], [0,7], [0,10], [0,11], [0,12], [0,13], [0,14], [0.0006647,1], [0.2881,8], [1.58,9] ]
chr1 10600 10800 id:4,qcat:[ [-0.02619,15], [0,1], [0,2], [0,3], [0,4], [0,6], [0,7], [0,8], [0,10], [0,11], [0,12], [0,13], [0,14], [0.1077,5], [0.4857,9] ]
This can be aggregated into multivec format:
clodius convert bedfile_to_multivec \
hg38/all.KL.bed.gz \
--assembly hg38 \
--starting-resolution 200 \
--row-infos-filename row_infos.txt \
--num-rows 15 \
--format epilogos
Other Data (multivec)¶
Multivec files are datatype agnostic. For use with generic data, create a segments file containing the maximum value for each segment. A segment is an arbitrary set of discontinuous blocks that the data is partitioned into. If the data has no natural grouping, one segment can be used which contains the maximum x value in the dataset:
segment1 20000
The individual datapoints should then be formatted as follows:
segment_name start end value
segment1 10 100 5
This can be converted to a multivec file using the following command:
clodius convert bedfile_to_multivec \
data.tsv \
--chromsizes-file segments.tsv \
--starting-resolution 1
The resulting output file can be ingested using higlass-manage:
higlass-manage.py ingest --filetype multivec --datatype multivec data.mv5